I nanoingegneri sviluppano un database predittivo per i materiali
Un algoritmo rivoluzionario espande lo spazio di esplorazione dei materiali di ordini di grandezza
I nanoingegneri della Jacobs School of Engineering dell’Università della California di San Diego hanno sviluppato un algoritmo di intelligenza artificiale che prevede la struttura e le proprietà dinamiche di qualsiasi materiale, esistente o nuovo, quasi istantaneamente. Conosciuto come M3GNet, l’algoritmo è stato utilizzato per sviluppare matterverse.ai , un database di oltre 31 milioni di materiali ancora da sintetizzare con proprietà previste da algoritmi di apprendimento automatico. Matterverse.ai facilita la scoperta di nuovi materiali tecnologici con proprietà eccezionali.
Il team dietro M3GNet, guidato dal professore di nanoingegneria della UC San Diego Shyue Ping Ong, utilizza matterverse.ai e le nuove funzionalità di M3GNet nella ricerca di elettrodi ed elettroliti più sicuri e ad alta densità energetica per batterie ricaricabili agli ioni di litio. Il progetto viene esplorato nel numero del 28 novembre della rivista Nature Computational Science .
Le proprietà di un materiale sono determinate dalla disposizione dei suoi atomi. Tuttavia, gli approcci esistenti per ottenere tale accordo sono proibitivi o inefficaci per molti elementi.
“Come per le proteine, dobbiamo conoscere la struttura di un materiale per prevederne le proprietà”. ha affermato Ong, direttore associato del Sustainable Power and Energy Center presso la Jacobs School of Engineering. “Ciò di cui abbiamo bisogno è un AlphaFold per i materiali”.
AlphaFold è un algoritmo AI sviluppato da Google DeepMind per prevedere la struttura delle proteine. Per costruire l’equivalente per i materiali, Ong e il suo team hanno combinato reti neurali a grafo con interazioni a molti corpi per creare un’architettura di deep learning che funzioni universalmente, con elevata precisione, su tutti gli elementi della tavola periodica.
“I grafici matematici sono rappresentazioni davvero naturali di una raccolta di atomi”, ha affermato Chi Chen, ex scienziato di progetto senior nel laboratorio di Ong e primo autore del lavoro, che ora è un architetto quantistico senior presso Microsoft Quantum. “Utilizzando i grafici, possiamo rappresentare l’intera complessità dei materiali senza essere soggetti all’esplosione combinatoria dei termini nei formalismi tradizionali”.
Per addestrare il proprio modello, il team ha utilizzato l’enorme database di energie, forze e sollecitazioni dei materiali raccolti nell’ultimo decennio nel progetto sui materiali . Il risultato è il potenziale interatomico M3GNet (IAP), che può prevedere le energie e le forze in qualsiasi insieme di atomi. Matterverse.ai è stato generato attraverso sostituzioni elementali combinatorie su oltre 5.000 prototipi strutturali nell’Inorganic Crystal Structure Database (ICSD). L’IAP M3GNet è stato quindi utilizzato per ottenere la struttura cristallina di equilibrio, un processo chiamato “rilassamento”, per la previsione delle proprietà.
Dei 31 milioni di materiali presenti oggi su matterverse.ai, si prevede che più di un milione siano potenzialmente stabili. Ong e il suo team intendono espandere notevolmente non solo il numero di materiali, ma anche il numero di proprietà previste da ML, comprese proprietà di alto valore con dimensioni di dati ridotte utilizzando un approccio multi-fedeltà che hanno sviluppato in precedenza .
Oltre ai rilassamenti strutturali, M3GNet IAP ha anche ampie applicazioni nelle simulazioni dinamiche di materiali e previsioni di proprietà.
“Ad esempio, siamo spesso interessati alla velocità con cui gli ioni di litio si diffondono in un elettrodo o elettrolita di una batteria agli ioni di litio. Più veloce è la diffusione, più velocemente puoi caricare o scaricare una batteria”, ha detto Ong. “Abbiamo dimostrato che M3GNet IAP può essere utilizzato per prevedere la conduttività del litio di un materiale con buona precisione. Crediamo veramente che l’architettura M3GNet sia uno strumento di trasformazione che può espandere notevolmente la nostra capacità di esplorare nuove chimiche e strutture dei materiali”.
Per promuovere l’uso di M3GNet, il team ha rilasciato il framework come codice Python open source su Github . Da quando ha pubblicato il preprint su Arxiv nel febbraio 2022, il team ha ricevuto l’interesse di ricercatori accademici e di quelli del settore. Ci sono piani per integrare M3GNet IAP come strumento in pacchetti di simulazione di materiali commerciali.
Questo lavoro è stato scritto da Chi Chen e Shyue Ping Ong della UC San Diego. La ricerca è stata finanziata principalmente dal Dipartimento dell’Energia degli Stati Uniti, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division nell’ambito del programma Materials Project. Parte del lavoro è stato finanziato da LG Energy Solution attraverso il Frontier Research Laboratory Program. Questo lavoro ha utilizzato l’Extreme Science and Engineering Discovery Environment (XSEDE).
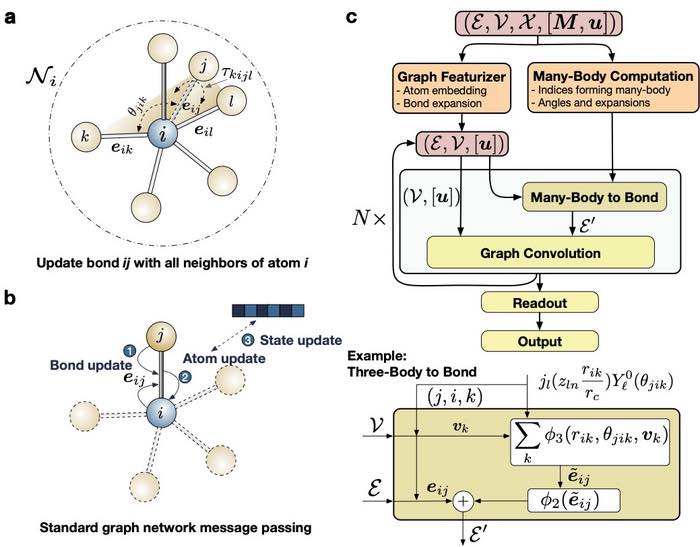
L’architettura del modello parte da un grafico con posizione inclusa, quindi passa attraverso un processo di caratterizzazione, seguito dai blocchi principali e dal modulo di lettura con output di energia, forza e sollecitazione.
Il processo di featurizzazione è costituito dal featurizzatore grafico e dal modulo di calcolo a molti corpi.
Nel featurizer grafico, il numero atomico degli elementi è stato incorporato in uno spazio di caratteristiche continue apprendibili e le distanze dei legami di coppia sono state espanse su una base impostata con valori e derivate fino al secondo ordine che vanno a zero al confine.
Il modulo di calcolo a molti corpi calcola gli indici degli atomi di interazione a tre corpi ea molti corpi e gli angoli associati.
Il blocco principale è costituito da due passaggi principali, vale a dire il modulo a molti corpi per legare e la convoluzione del grafico standard.
La fase da molti corpi a legame calcola la nuova informazione di legame eij considerando l’intero ambiente di legame Ni dell’atomo i tramite angoli a molti corpi come θjik, τkijl, ecc., e la lunghezza del legame rik,rij,ril, ecc. la convoluzione del grafico standard aggiorna il legame, l’atomo e le informazioni sullo stato opzionale in modo iterativo.
Durante la fase di lettura, le informazioni sugli atomi nel grafico sono state passate a un MLP con gate per ottenere l’energia atomica, che si somma all’energia totale.
Le derivate dell’energia totale danno uscite di forza e sollecitazione.
che si somma all’energia totale.
Le derivate dell’energia totale danno uscite di forza e sollecitazione.
che si somma all’energia totale.
Le derivate dell’energia totale danno uscite di forza e sollecitazione.
CREDITO
Università della California San Diego